I love doing simulations because they offer a powerful playground for exploring complex physical and chemical problems. In particular, I use computational tools and theoretical models to investigate chemical reactions and emergent quantum phenomena, including those result from strong light–matter interactions.
The figure that chatGPT generated according to the paragraph above. Obviously it is not familiar with Schrödinger equation and Maxwell's equations.
Many of these emergent quantum phenomena involve nonadiabatic dynamics, where the electrons are strongly coupled with the nuclei (breaking down the Born–Oppenheimer approximation); or the matters are strongly coupled to the optical fields (requiring a quantum treatment of the field via second quantization). To address these challenges in a computationally tractable way, I focus on mixed quantum–classical methods as an alternative to fully quantum mechanical simulations.
Some of the broad research questions that guide my work include:
How accurately can classical mechanics capture quantum features and observables?
Can we develop more efficient and accurate methods for modeling nonadiabatic dynamics?
How can we combine theory with experimental data (e.g., spectra) to understand nonadiabatic processes in realistic, complex environments?
Can we design or control nonadiabatic phenomena based on the theoretical insights we gain?
The following projects reflect my efforts in tackling these questions. While they may not fully (or even partially) answer them, each project addresses its own research question as well.
Strong light–matter interactions
Strong light–matter interactions have been shown to manipulate and engineer the physical and chemical properties of molecules and materials, but the underlying mechanisms are still not well understood. Within a weak coupling regime, people typically used steady classical light to couple with the matter and focus only on the matter part. However, a classical description of light cannot capture quantum phenomena such as spontaneous emission, which arises from vacuum fluctuations, as well as the quantitative agreement with quantum exact results when the photon quantum number is small, or the light–matter interactions are strong.
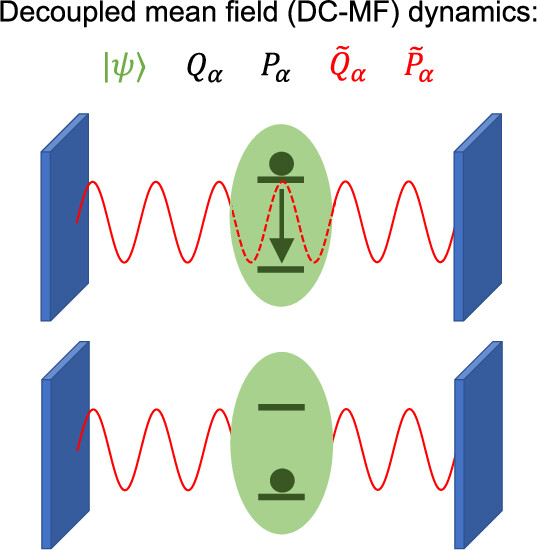
To address this, a quantum discription of optical field (incorporating quantum electrodynamics), although computationally expensive, are neccessary. To balance accuracy and efficiency, we exlpoited existing nonadiabatic dynamics models in the theoretical chemistry community and modified the Ehrenfest (mean-field) model to include some of the quantum characters of optical field. The new model we developed is named Decoupled Mean-field (DC-MF).
Decoupled Mean-Field (DC-MF) model: an efficient and accurate model for strong light–matter interactions
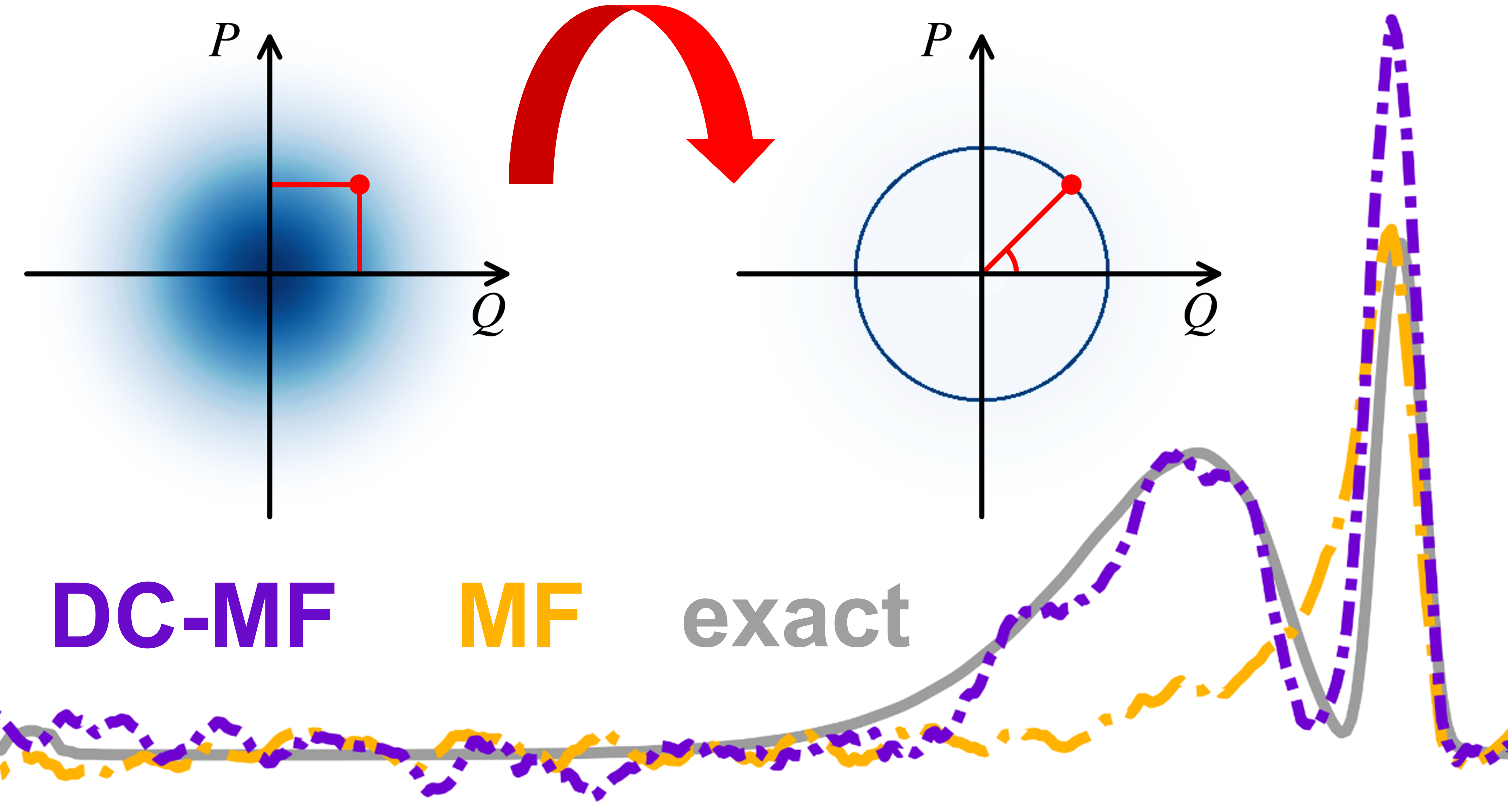
The key idea behind the DC-MF model is to separate the optical field into vacuum and thermal fluctuations, and to decouple the vacuum flucuations from the ground state.1 We came up with this “decoupling scheme” in order to address the issue of the standard Ehrenfest dynamics that results in unphysical energy transfer from zero-temperature vacuum fluctuations to the ground-state emitter (a zero-point energy leakage of the optical field). Beyond resolving this issue, our model also yields significant improvement in accuracy and good alignment with exact quantum reference.
We also invoked so-called focused sampling to initiate the vacuum fluctuations (only sampled phase-space variables that give zero-point energy) instead of the commonly used Wigner quasiprobability distribution. This not only reduces the number of required trajectories but also extends the applicability of the DC-MF model to a wider range of quantumness.2
The great computational scalability renders the DC-MF model applicable and feasible for various light–matter phenomena. We are currently integrating DC-MF with finite-difference time-domain (FDTD) method, a prevalent method for simulating nano-optics, to study complex light–matter interactions in realistic nanophotonic conditions.
Left: idea of DC-MF.1 Right: great agreement between DC-MF and exact quantum reference (CISD).1
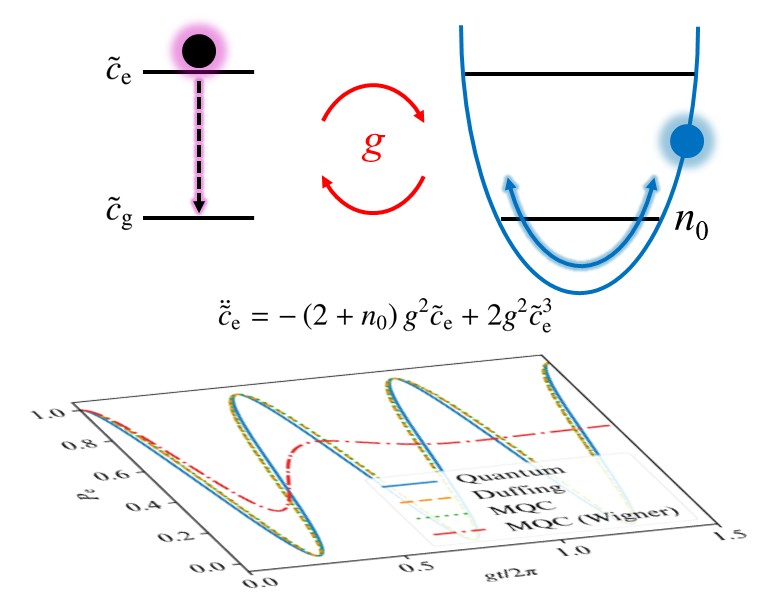
We analytically derived a differential equation governing mixed quantum–classical dynamics for the Rabi problem.3 We showed that the resulting dynamics are described by an unforced and undamped Duffing equation, yielding anharmonic osciilations. Furthermore, we demonstrate that to reproduce the quantum results, the classical optical field needs to be initialized in around but slightly above the zero-point energy. This work provides guidance in the application of mixed quantum–classical dynamics to classes of problems involving small quantum numbers.
The dynamics governed by Duffing equation, yielding anharmonic oscillations.3
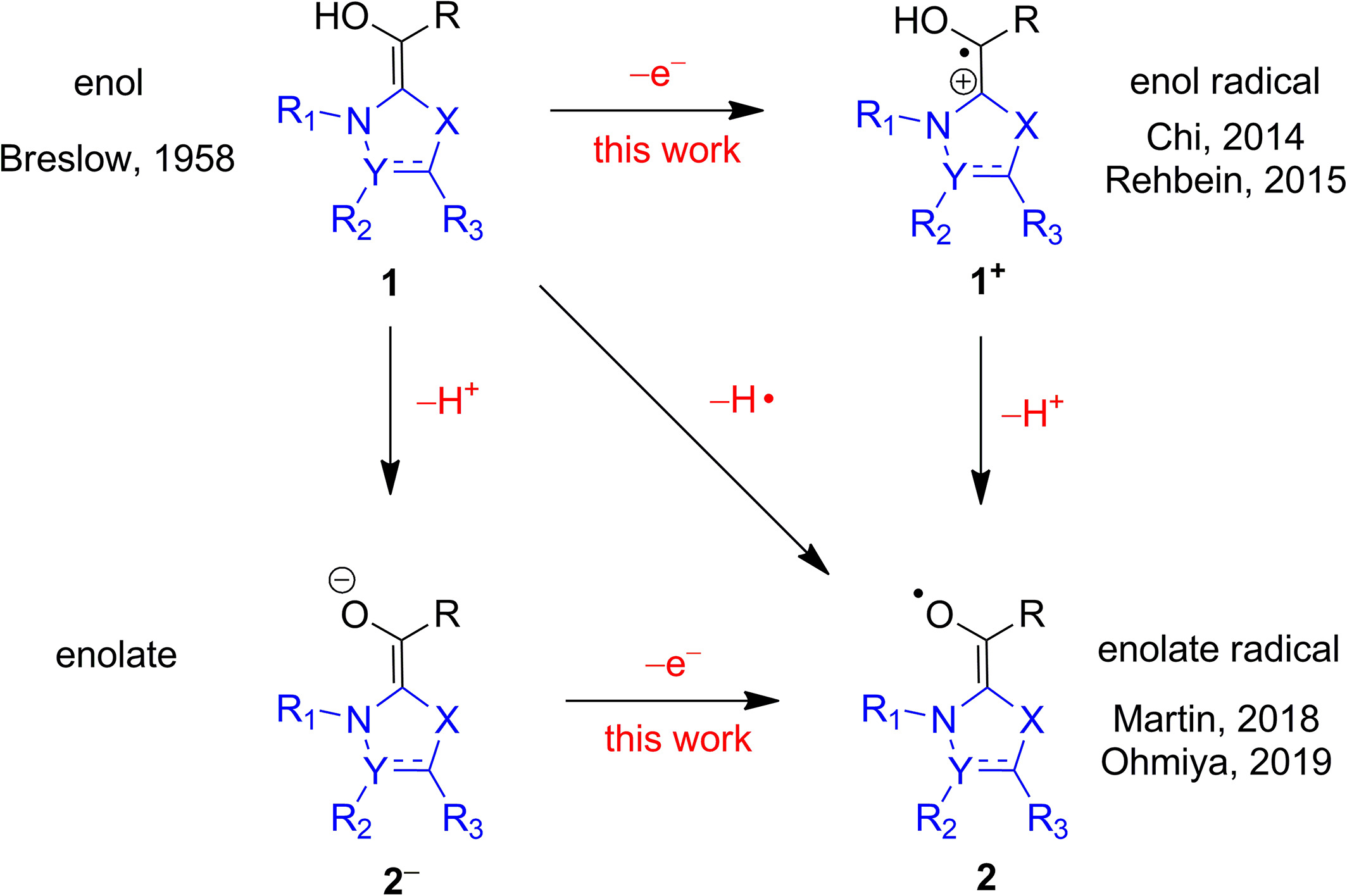
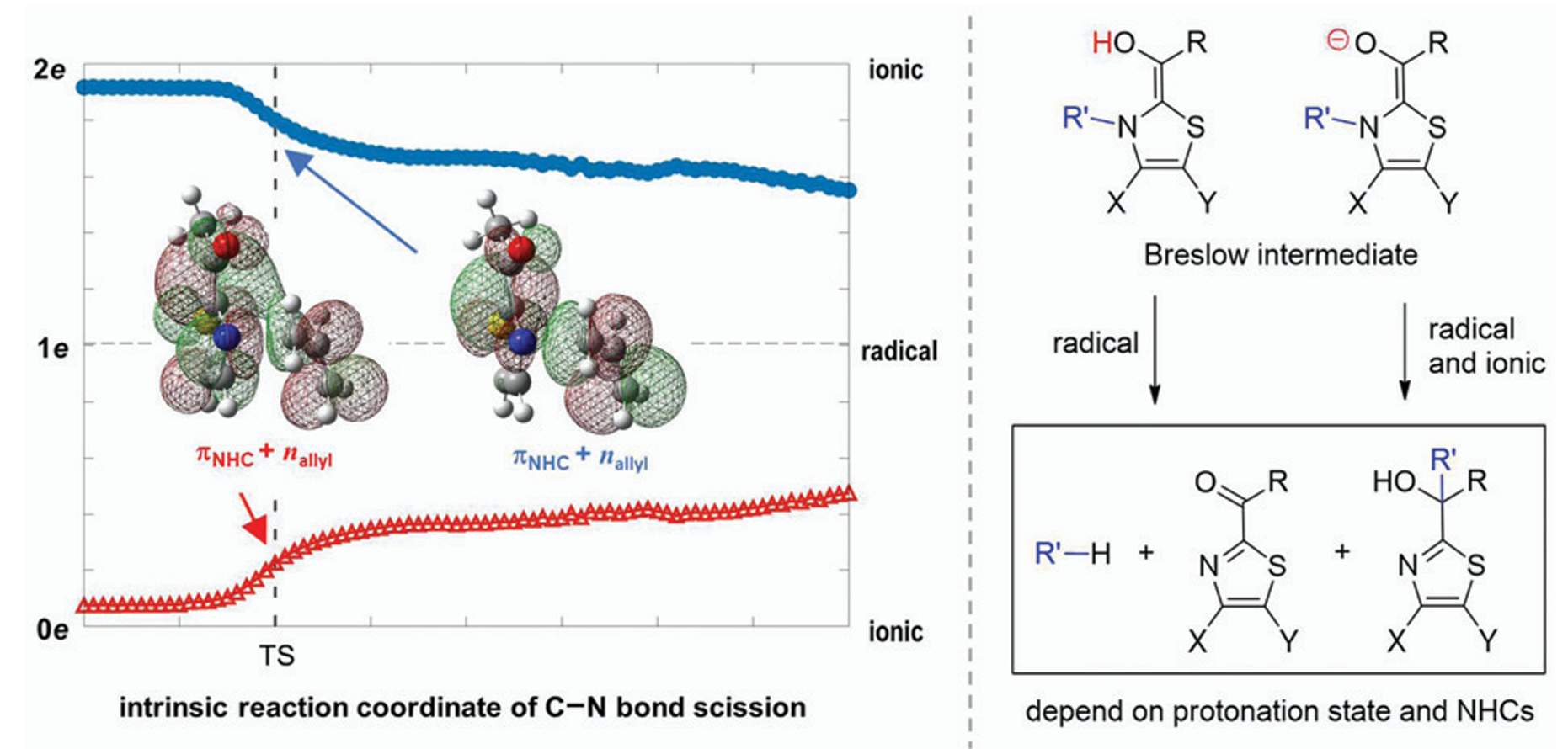
Radical mechanisms of Breslow intermediates in NHC-catalyzed reactions
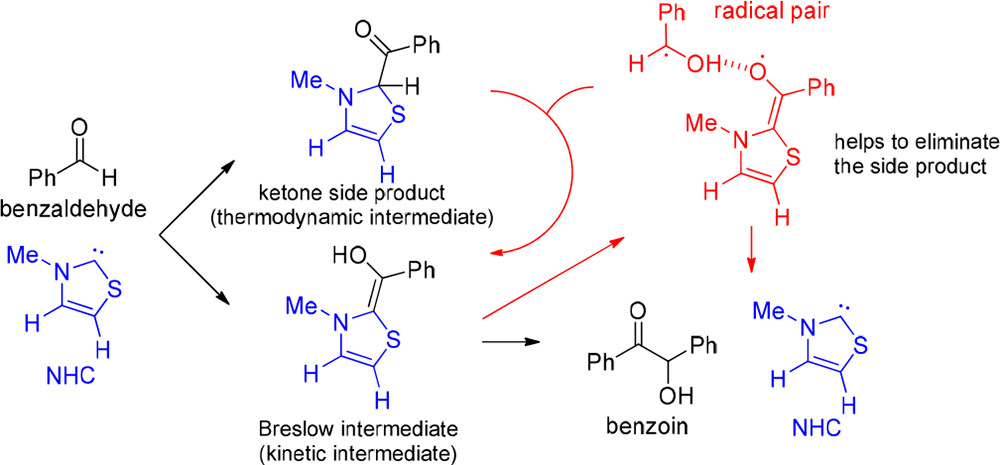
We found that the enolate form of Breslow intermediates can produce radicals through electron removal from a metastable dipole-bound state.4 In the case of benzoin condensation, the hydrogen-atom-transfer mechanism has a high energy barrier, but the resulting radicals may play a role to eliminate the side products.5 These findings align with experimental observations.
We also explored the radical mechanism in the fragmentation and rearrangement of Breslow intermediates and showed that the radical and ionic fragmentation pathways can coexist. The dominant pathway depends on the chemical structures and the protonation states of Breslow intermeidates.6
Left: dipole-bound orbital of a Breslow intermediate.4 Middle: energy surfaces of singlet, dipole-bound, and doublet states.4 Right: Hydrogen-atom transfer pathway for generating radicals.5
References
A Mean-Field Treatment of Vacuum Fluctuations in Strong Light–Matter Coupling
Mean-field mixed quantum–classical dynamics could provide a much-needed means to inexpensively model quantum electrodynamical phenomena by describing the optical field and its vacuum fluctuations classically. However, this approach is known to suffer from an unphysical transfer of energy out of the vacuum fluctuations when the light–matter coupling becomes strong. We highlight this issue for the case of an atom in an optical cavity and resolve it by introducing an additional set of classical coordinates to specifically represent vacuum fluctuations whose light–matter interaction is scaled by the instantaneous ground-state population of the atom. This not only rigorously prevents the aforementioned unphysical energy transfer but is also shown to yield a radically improved accuracy in terms of the atomic population and the optical field dynamics, generating results in excellent agreement with full quantum calculations. As such, the resulting method emerges as an attractive solution for the affordable modeling of strong light–matter coupling phenomena involving macroscopic numbers of optical modes.
Focused Sampling for Low-Cost and Accurate Ehrenfest Modeling of Cavity Quantum Electrodynamics
An economic modeling approach for cavity quantum electrodynamics is provided by mean-field dynamics, wherein the optical field is described classically while a self-consistent interaction with quantum emitters is incorporated through the Ehrenfest theorem. However, conventional implementations of mean-field dynamics are known to suffer from a catastrophic leakage of zero-point energy, to lose accuracy in the short-cavity limit, and to require large numbers of trajectories to be sampled. Here, we address these three shortcomings within a single integrated approach. This approach builds on our recently-proposed modification of the Ehrenfest theorem, referred to as decoupled mean-field (DC-MF) dynamics, in combination with a focused sampling scheme that enforces zero-point energy at the single-trajectory level. The approach is shown to yield high accuracy in both short and long-cavity limits while reaching convergence within a minimal amount of trajectories.
This paper has been selsected as Cover and in the 2025 JCP Emerging Investigators Special Collection of the Journal of Chemical Physics, Volume 162, Issue 22, June 2025.
We apply a mixed quantum–classical (MQC) approach to the quantum Rabi model, involving a classical optical field coupled self-consistently to a quantum two-level system. Under the rotating wave approximation, we analytically show that this approach yields persistent yet anharmonic Rabi oscillations, governed by an undamped and unforced Duffing equation. We consider the single-quantum limit, where we find that such anharmonic Rabi oscillations closely follow full-quantum results once zero-point energy is approximately enforced when initializing the optical field coordinate. Our findings provide guidance in the application of MQC dynamics to classes of problems involving small quantum numbers and far away from decoherence.
Dipole-Bound States and Substituent Effects of Breslow Intermediates in the Enolate Form
Ming-Hsiu Hsieh, Gou-Tao Huang, and Jen-Shiang K. Yu
Breslow intermediates play crucial roles in both umpolung and redox reactions in N-heterocyclic carbene catalysis. Compared to the well-known nucleophilic character, the electronic structure of Breslow intermediates on the radical route is still unclear. We investigate the potential energy surfaces with high-level ab initio methods for four typical Breslow intermediates in both of their enol and enolate forms. In the enol form, high energies of around 60 kcal/mol to the Rydberg-like states and those higher than 120 kcal/mol to remove an electron demonstrate that the enol Breslow intermediates tend not to generate radicals unless strong oxidants are present. The low-lying dipole-bound states and small electron detachment energies in the enolate form in contrast show that the enolate Breslow intermediates are possible precursors to radicals. More importantly, metastable dipole-bound states exist in the imidazole- and the triazole-based enolate Breslow intermediates. Energies to detach one electron of several enolate Breslow intermediates reveal that the bulky and electron-withdrawing groups stabilize the singlet ground states, which explains that the utilization of such substituents can lead to successful isolation for Breslow intermediates in experiments.
Can the Radical Channel Contribute to the Catalytic Cycle of N-Heterocyclic Carbene in Benzoin Condensation?
Ming-Hsiu Hsieh, Gou-Tao Huang, and Jen-Shiang K. Yu
NHC can catalyze benzoin condensation via the key Breslow intermediate. EPR spectroscopy recently confirmed the existence of the radical species, but its catalytic role is still unclear. Herein, we use density functional approaches to study the radical-associated pathway in comparison with the nonradical mechanism reported previously. Theoretical investigations show that the nonradical path (∆G⧧ = 18.7 kcal/mol) is more kinetically favorable than the radical route (∆G⧧ = 27.6 kcal/mol), which is initialized by the hydrogen abstraction from the Breslow intermediate by benzaldehyde, leading to a radical pair. The product formation is thus dominated by the nonradical pathway. In addition, the Breslow intermediate is less stable than its keto form, which blocks the benzoin condensation, and the radical species could play an important role in assisting the tautomerization and promoting the catalytic reaction.
Fragmentation and Rearrangement of Breslow Intermediates: Branches to Both Radical and Ionic Pathways
Breslow intermediates are the key species in N-heterocyclic carbene-catalyzed reactions to promote the C–C bond formation. As the fragmentation and rearrangement of Breslow intermediates terminate the catalytic cycle of N-heterocyclic carbene, two mechanisms under debate have been proposed in terms of the radical channel and the ionic route. Theoretical calculations demonstrate herein that ionic and radical characteristics can coexist, depending on the protonation state of the hydroxyl group in Breslow intermediates: radicals are merely generated in the enol system, while both ionic and radical species exist in the enolate system with a lower barrier. Complete pathways for thiamin analogue and N-allyl benzothiazole Breslow intermediates are exclusively constructed considering experimental conditions. The growing population of the enolate under higher pH values rationalizes the increased rate of the fragmentation of thiamin. The fragmentation products of thiamin, namely pyrimidine and ketone, are the thermodynamic products, while the tertiary alcohol is both the kinetic and thermodynamic product for N-allyl benzothiazole Breslow intermediate via a Claisen-like rearrangement. Other NHCs used to synthesize tertiary alcohols could form the enolate due to the base, followed by the production of stable radicals and recombination to form tertiary alcohols. It is concluded that specific protonation states and chemical structures of NHCs account for the distinct mechanisms.







 Mixed quantum–classical dynamics yields anharmonic Rabi oscillationsJ. Chem. Phys. 2025, 162, 224109
Mixed quantum–classical dynamics yields anharmonic Rabi oscillationsJ. Chem. Phys. 2025, 162, 224109